¿Para que sirve Saxenda 6 mg / ml x 3 dispositivos prellenados?
Saxenda es un medicamento para perder peso que contiene el principio activo liraglutida. Es parecido a una hormona de origen natural llamada péptido-1 similar al glucagón (GLP-1) que se libera en el intestino después de comer. Saxenda actúa sobre los receptores del cerebro que controlan el apetito y le hacen sentirse más lleno y menos hambriento. De esta forma, puede ayudarle a comer menos y reducir su peso corporal.
- Composición: Un ml de solución contiene 6 mg de Liraglutida*. Un dispositivo prellenado contiene 18 mg de Liraglutida en 3 ml. * Análogo humano del Péptido-1 similar al Glucagón (GLP-1) producido por tecnología de ADN recombinante en Saccharomyces cerevisiae. Para consultar la lista completa de excipientes, ver Lista de excipientes. Forma farmacéutica: Solución inyectable. Solución transparente, incolora o casi incolora e isotónica; pH=8.15. Lista de excipientes: Fosfato Disódico Dihidratado, Propilenglicol, Fenol, Acido Clorhídrico (para ajuste del pH), Hidróxido de Sodio (para ajuste del pH), Agua para Preparaciones Inyectables.
- Acción Terapéutica: Grupo farmacoterapéutico: fármacos utilizados en la diabetes, otros fármacos hipoglicemiantes, excluyendo las insulinas. Código ATC: A10BX07.
- Indicaciones: Saxenda® está indicado, en combinación con una dieta baja en calorías y un aumento de la actividad física, para controlar el peso en pacientes adultos con un índice de masa corporal (IMC) inicial de ≥ 30 kg/m2 (obesidad) o ≥ 27 kg/m2 a < 30 kg/m2 (sobrepeso) que presentan al menos una comorbilidad relacionada con el peso, como alteraciones de la glicemia (prediabetes o diabetes mellitus tipo 2), hipertensión, dislipidemia o apnea obstructiva del sueño. El tratamiento con una dosis diaria de 3.0 mg de Saxenda® se debe interrumpir si después de 12 semanas los pacientes no han perdido al menos el 5% de su peso corporal inicial.
- Propiedades: Propiedades farmacodinámicas: Mecanismo de acción: Liraglutida es un análogo acilado humano del péptido-1 similar al glucagón (GLP-1) con un 97% de homología de secuencia de aminoácidos con el GLP- 1 humano endógeno. Liraglutida se une al receptor de GLP-1 (GLP-1R) y lo activa. El GLP-1 es un regulador fisiológico del apetito y de la ingesta de alimentos, pero el mecanismo exacto de acción no está completamente claro. En estudios llevados a cabo con animales, la administración periférica de liraglutida supuso la absorción en regiones específicas del cerebro implicadas en la regulación del apetito, donde liraglutida, a través de la activación específica de GLP-1R, aumentó las señales de saciedad básicas y redujo las señales de hambre básicas que permitieron perder peso. Efectos farmacodinámicos: Liraglutida contribuye a reducir el peso corporal en humanos principalmente a través de la pérdida de materia grasa con reducciones relativas de grasa visceral que son mayores que la pérdida de grasa subcutánea. Liraglutida regula el apetito porque aumenta la sensación de plenitud y saciedad, a la vez que reduce la sensación de hambre y el posible consumo de alimentos, lo que conduce a una reducción en la ingesta de alimentos. En comparación con el placebo, liraglutida no incrementa el gasto de energía. Liraglutida estimula la secreción de insulina y disminuye la secreción de glucagón de un modo dependiente de la glucosa, lo que reduce la glucosa en ayunas y postprandial. El efecto hipoglicemiante es mayor en pacientes con prediabetes y diabetes que en los pacientes con normoglicemia. Los ensayos clínicos sugieren que liraglutida mejora y mantiene la función de las células beta según HOMA-B y la relación proinsulina/insulina. Eficacia y seguridad clínica: La eficacia y seguridad de liraglutida para controlar el peso en combinación con una menor ingesta de calorías y un aumento de la actividad física se evaluaron en 4 ensayos aleatorizados de fase 3, doble ciegos y controlados por placebo en los que participaron un total de 5.358 pacientes. Ensayo 1 (SCALE Obesity & Pre-Diabetes - 1839): Ensayo de 56 semanas de duración en el que se evaluó la pérdida de peso corporal en 3.731 pacientes aleatorizados (de los cuales 2.590 completaron el ensayo) con obesidad y con sobrepeso con una de las siguientes patologías: prediabetes, hipertensión o dislipidemia. El 61% de los pacientes tenía prediabetes al inicio. Ensayo 2 (SCALE Diabetes - 1922): Ensayo de 56 semanas de duración en el que se evaluó la pérdida de peso corporal en 846 pacientes aleatorizados (de los cuales 628 completaron el ensayo) con obesidad y sobrepeso además de diabetes mellitus tipo 2 insuficientemente controlada (rango de HbA1c 7-10%). El tratamiento al comienzo del ensayo consistía solamente en dieta y ejercicio, metformina, una sulfonilurea, una glitazona como monoterapias, o una combinación de estos. Ensayo 3 (SCALE Sleep Apnoea - 3970): Ensayo de 32 semanas de duración en el que se evaluó la gravedad de la apnea del sueño y la pérdida de peso corporal en 359 pacientes aleatorizados (de los cuales 276 completaron el ensayo) con obesidad y apnea del sueño obstructiva moderada o grave. Ensayo 4 (SCALE Maintenance - 1923): Ensayo de 56 semanas de duración en el que se evaluaron el mantenimiento y la pérdida de peso corporal en 422 pacientes aleatorizados (de los cuales 305 completaron el ensayo) con obesidad y sobrepeso además de hipertensión o dislipidemia tras experimentar una pérdida de peso anterior del ≥5% a causa de una dieta baja en calorías. Peso corporal: Se alcanzó una mayor pérdida de peso con liraglutida que con placebo en pacientes obesos / con sobrepeso en todos los grupos estudiados. En todas las poblaciones del ensayo, fue mayor el porcentaje de pacientes que experimentaron una pérdida de peso ≥5% y >10% con liraglutida en comparación con placebo. En el ensayo 1: La media del cambio desde el peso corporal inicial a la semana 56 fue de -8.0% (-8.4 kg) para liraglutida en comparación a -2.6% (-2.8 kg) para placebo (diferencia estimada en el tratamiento [sigla ETD, en inglés] media del cambio en %): -5.4 [95% CI -5.8; -5.0], p<0.0001, ETD (media del cambio en kg): -5.6 [95%IC -6.0; -5.1], p<0.0001). La proporción de pacientes con pérdida de peso del 5% y 10% a la semana 56 fue del 63.5% y 32.8% respectivamente para liraglutida en comparación al 26.6% y 10.1% respectivamente para placebo, relación de oportunidades estimada (de pérdida de ≥5% de peso corporal): 4.8 [95%IC 4.1; 5.6], p<0.0001, relación de oportunidades estimada (de pérdida de >10% de peso corporal): 4.3 [95%IC 3.5; 5.3], p<0.0001). En el ensayo 2: La media del cambio desde el peso corporal inicial a la semana 56 fue de -5.9% (-6.2 kg) para liraglutida en comparación a -2.0% (-2.2 kg) para placebo (diferencia estimada en el tratamiento (ETD) (media del cambio en %): -4.0 [95%IC -4.8; -3.1], p<0.0001, ETD (media del cambio en kg): -4.1[95%IC -5.0; -3.1], p<0.0001). La proporción de pacientes con pérdida de peso del 5% y 10% a la semana 56 fue del 49.8% y 22.9% respectivamente para liraglutida en comparación al 13.5% y 4.2% respectivamente para placebo, relación de oportunidades estimada (de pérdida de ≥5% de peso corporal): 6.4[95%IC 4.1; 10.0], p<0.0001, relación de oportunidades estimada (de pérdida de >10% de peso corporal): 6.8 [95%IC 3.4; 13.8], p<0.0001). En el ensayo 3: La media del cambio desde el peso corporal inicial a la semana 32 fue de -5.7% (-6.8 kg) para liraglutida en comparación a -1.6% (-1.8 kg) para placebo (diferencia estimada en el tratamiento (ETD) (media del cambio en %): -4.2[95%IC -5.2; -3.1], p<0.0001, ETD (media del cambio en kg): -4.9[95%IC -6.2; -3.7], p<0.0001). La proporción de pacientes con pérdida de peso del 5% a la semana 32 fue del 46.4% para liraglutida en comparación al 18.1% para placebo, relación de oportunidades estimada 3.9[95%IC 2.4; 6.4], p<0.0001). En el ensayo 4: Fue mayor el número de pacientes que mantuvieron la pérdida de peso alcanzada con anterioridad al inicio del tratamiento con liraglutida que con placebo (81.4% y 48.9%, respectivamente). La media del cambio desde el peso corporal inicial a la semana 56 fue del -6.3% (-6.0 kg) para liraglutida en comparación a -0.2% (-0.2 kg) para placebo (diferencia estimada en el tratamiento (ETD) (media del cambio en %): - 6.1[95%IC -7,5; -4,6], p<0,0001, ETD (media del cambio en kg): -5.9 [95%IC -7.3; -4.4], p<0.0001). La proporción de pacientes con pérdida de peso del 5% y 10% a la semana 56 fue del 50.7% y 27.4% respectivamente para liraglutida en comparación al 21.3% and 6.8% respectivamente para placebo, relación de oportunidades estimada (de pérdida de ≥5% de peso corporal): 3.8[95%IC 2.4; 6.0], p<0.0001, relación de oportunidades estimada (de pérdida de >10% de peso corporal): 5.1[95%IC 2.7 ; 9.7], p<0.0001). Los datos sobre la pérdida de peso, la evolución temporal y la distribución acumulada del cambio de peso (en %) se presentan en las figuras 1, 2, y 3. Pérdida de peso después de 12 semanas de tratamiento con liraglutida (3.0 mg): Los pacientes con respuesta inicial rápida se definieron como los pacientes en los que se produjo una pérdida de peso ≥5% tras 12 semanas con la dosis de tratamiento de liraglutida (4 semanas de aumento de dosis y 12 semanas con la dosis de tratamiento). En el ensayo 1: El 67.5% de los pacientes lograron una pérdida de peso ≥5% después de 12 semanas. En el ensayo 2: El 50.4% de los pacientes lograron una pérdida de peso ≥5% después de 12 semanas. Si se continúa el tratamiento con liraglutida, se prevé que el 86.2% de los pacientes con respuesta inicial rápida alcancen una pérdida de peso ≥5% y que el 51% alcance una pérdida de peso ≥10% después de 1 año de tratamiento. Se prevé que los pacientes con respuesta inicial rápida que completen 1 año de tratamiento pierdan una media del 11.2% de su peso corporal inicial (9.7% en hombres y 11.6% en mujeres). Para los pacientes que presentan una pérdida de peso <5% tras 12 semanas con la dosis de tratamiento de liraglutida, la proporción de pacientes que no consiguen experimentar una pérdida de peso ≥10% tras 1 año es del 93.4%. Control glicémico: El tratamiento con liraglutida mejora de forma significativa los parámetros glicémicos en subpoblaciones con normoglicemia, prediabetes y diabetes mellitus tipo 2. En el ensayo 1: Desarrollaron menos diabetes mellitus tipo 2 los pacientes tratados con liraglutida que los tratados con placebo (0.2% frente a 1.1%). En comparación con los pacientes tratados con placebo, la prediabetes inicial desapareció en más pacientes (69.2% frente a 32.7%). Desde una HbA1c inicial de 5.6%, los pacientes en el grupo de liraglutida tuvieron una media de reducción de HbA1c del -0.3% a la semana 56 en comparación a -0.1% en el grupo placebo (ETD: -0.23[95%IC -0.25; -0.21], p<0.0001. Desde una GPA inicial de 5.3 mmol/l, los pacientes en el grupo de liraglutida tuvieron una media de reducción de GPA del -0.4 mmol/l a la semana 56 en comparación a -0.01 mmol/l en el grupo placebo (ETD: -0.38 [95%IC -0.42; -0.35], p<0.0001). En el ensayo 2: Desde una HbA1c inicial de 7.9%, los pacientes en el grupo de liraglutida tuvieron una media de reducción de HbA1c del -1.3% a la semana 56 en comparación a -0.4% en el grupo placebo (ETD: -0.9[95%IC -1.1; -0.8], p<0.0001). Desde una GPA inicial de 8.8 mmol/l en el grupo de liraglutida y 8.6 mmol/l en el grupo placebo, los pacientes tuvieron una media de reducción de GPA del -1.9 mmol/l y -0.1 mmol/l a la semana 56, respectivamente (ETD: -1.8[95%IC - 2.1; -1.4], p<0.0001). Factores de riesgo cardiometabólicos: El tratamiento con liraglutida mejoró de forma significativa la presión arterial sistólica (sigla SBP, en inglés) y la circunferencia de la cintura en comparación con placebo. En el ensayo 1: Desde un SBP inicial de 123.0 mmHg en el grupo de liraglutida y 123.3 mmHg en el grupo placebo, los pacientes tuvieron una media de reducción de -4.3 mmHg y -1.5 mmHg a la semana 56, respectivamente (ETD: -2.8[95%IC -3.6; -2.1], p<0.0001). Desde una presión arterial sistólica (SBP) inicial de 78.7 mmHg en el grupo de liraglutida y 78.9 mmHg en el grupo placebo, los pacientes tuvieron una media de reducción de DBP de -2.7 mmHg y -1.8 mmHg a la semana 56, respectivamente (ETD: -0.9[95%IC -1.4; -0.4], p<0.05). Desde una circunferencia de la cintura inicial de 115.0 cm en el grupo de liraglutida y 114.5 cm en el grupo placebo, los pacientes tuvieron una media de reducción de -8.2 cm y -4.0 cm a la semana 56, respectivamente (ETD: - 4.2[95%IC -4.7; -3.7], p<0.0001). En el ensayo 2: Desde una SBP inicial de 128.9 mmHg en el grupo de liraglutida y 129.2 mmHg en el grupo placebo, los pacientes tuvieron una media de reducción de SBP de -3.0 mmHg y -0.4 mmHg a la semana 56, respectivamente (ETD: -2.6[95%IC -4.6; -0.6], p<0.0001). Desde una presión arterial sistólica (DBP) inicial de 79 mmHg en el grupo de liraglutida y 79.3 mmHg en el grupo placebo, los pacientes tuvieron una media de reducción de DBP de -1.0 mmHg y -0.6 mmHg a la semana 56, respectivamente (ETD: -0.4[95%IC -1.7; 1.0], p=0.5918). Desde una circunferencia de la cintura inicial de 118.1 cm en el grupo de liraglutida y 117.3 cm en el grupo placebo, los pacientes tuvieron una media de reducción de -6.0 cm y -2.8 cm a la semana 56, respectivamente (ETD: -3.2[95%IC -4.2; -2.2], p<0.0001). Índice de Apnea-Hipopnea (sigla AHI, en inglés): El tratamiento con liraglutida reduce de manera significativa la gravedad de la apnea obstructiva del sueño como indica el cambio con respecto al nivel inicial del AHI frente a placebo (-12.2 eventos/hora para liraglutida en comparación a -6.1 eventos/hora para placebo, ETD: -6.1[95%IC -11.0; -1.2], p<0.05).
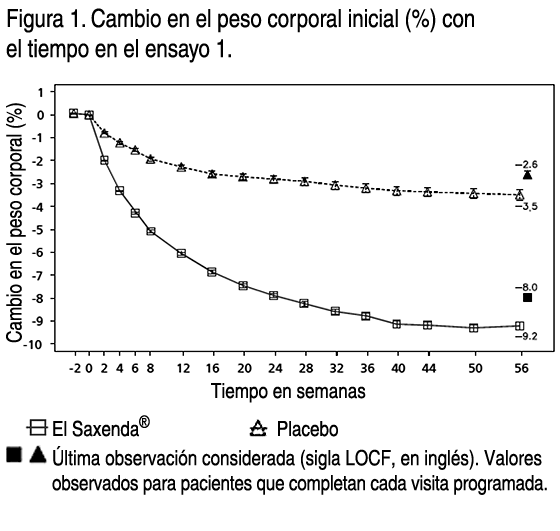
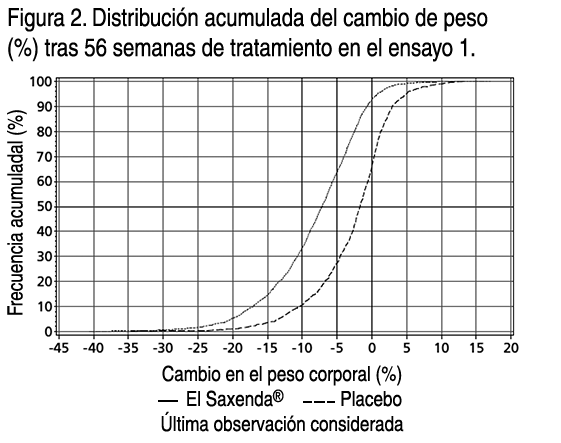
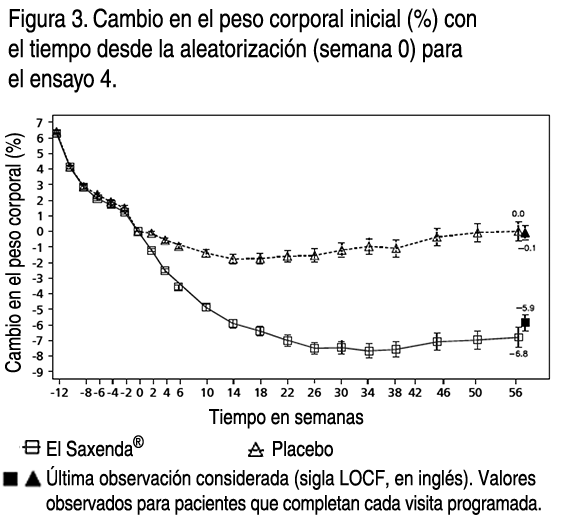
- Antes de la semana 0, el tratamiento de los pacientes consistía exclusivamente en una dieta baja en calorías y ejercicio. En la semana 0 los pacientes fueron aleatorizados para recibir Saxenda® o placebo. Inmunogenicidad: De acuerdo con las propiedades potencialmente inmunogénicas de los fármacos que contienen proteínas o péptidos, los pacientes pueden desarrollar anticuerpos antiliraglutida tras el tratamiento con liraglutida. En los ensayos clínicos, el 2.5% de los pacientes tratados con liraglutida desarrolló anticuerpos antiliraglutida. La formación de anticuerpos no se ha asociado con una reducción en la eficacia de liraglutida. Evaluación cardiovascular: Un grupo de expertos independiente y externo se encargó de valorar los acontecimientos adversos cardiovasculares graves (sigla MACE en inglés) y los definió como infarto de miocardio no mortal, accidente cerebrovascular no mortal y muerte cardiovascular. En todos los ensayos clínicos a largo plazo realizados con Saxenda®, se produjeron 6 MACE en pacientes tratados con liraglutida y 10 MACE en pacientes que recibieron placebo. La razón de riesgo y el IC 95% es 0.33 [0,12; 0.90] para liraglutida en comparación con placebo. En los ensayos clínicos de fase 3 se ha observado que liraglutida produce un aumento medio de la frecuencia cardíaca desde el valor inicial de 2.5 latidos por minuto (el valor varía entre los ensayos de 1.6 a 3.6 latidos por minuto). La frecuencia cardíaca alcanzó su valor máximo después de 6 semanas aproximadamente. No se ha determinado el impacto clínico a largo plazo de este aumento medio de la frecuencia cardíaca. Este cambio en la frecuencia cardíaca fue reversible tras la interrupción del tratamiento con liraglutida (ver Advertencias y precauciones especiales de empleo). Propiedades farmacocinéticas: Absorción: La absorción de liraglutida tras la administración por vía subcutánea fue lenta, alcanzando su concentración máxima aproximadamente a las 11 horas tras su administración. La media de la concentración en estado estacionario de liraglutida (ABCT/24) alcanzó aproximadamente los 31 nmol/l en pacientes obesos (IMC 30-40 kg/m2) tras la administración de 3 mg de liraglutida. La exposición a liraglutida aumentó proporcionalmente con la dosis. La biodisponibilidad absoluta de liraglutida tras su administración por vía subcutánea es de aproximadamente un 55%. Distribución: El volumen de distribución aparente medio tras la administración subcutánea es de 20-25 l (para una persona que pesa unos 100 kg). Liraglutida se encuentra ampliamente ligada a proteínas plasmáticas (>98%). Biotransformación: Durante 24 horas tras la administración de una única dosis de [3H]- liraglutida a sujetos sanos, el componente mayoritario en plasma fue liraglutida intacta. Se detectaron 2 metabolitos minoritarios en el plasma (≤9% y ≤5% de la exposición a radioactividad plasmática total). Eliminación: Liraglutida se metaboliza endógenamente de un modo similar al de las grandes proteínas sin un órgano específico como ruta principal de eliminación. Tras una dosis de [3H]-liraglutida, no se detectó liraglutida intacta en orina o heces. Únicamente una proporción menor de la radioactividad administrada se excretó en forma de metabolitos relacionados con liraglutida a través de orina o heces (6% y 5%, respectivamente). La radioactividad en orina y heces se excretó principalmente durante los primeros 6-8 días y correspondió a 3 metabolitos minoritarios, respectivamente. El clearance medio tras la administración por vía subcutánea de liraglutida es de aproximadamente 0.9-1.4 l/h con una vida media de eliminación de aproximadamente 13 horas. Poblaciones especiales: Pacientes de edad avanzada: La edad no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de liraglutida según los resultados de un análisis de datos farmacocinéticos de la población de pacientes obesos y con sobrepeso (entre 18 y 82 años). No es necesario un ajuste de dosis en función de la edad. Sexo: Según los resultados de los análisis farmacocinéticos de la población, las mujeres tienen un clearance de liraglutida ajustado al peso que es inferior en un 24% al de los hombres. Según los datos de respuesta a la exposición, no es necesario un ajuste de dosis en función del sexo del paciente. Origen étnico: El origen étnico no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de liraglutida según los resultados del análisis farmacocinético de la población en el que se incluyeron pacientes obesos y con sobrepeso de grupos de población blanca, negra, asiática, hispanoamericana y no hispanoamericana. Peso corporal: La exposición a liraglutida disminuye con un aumento del peso corporal inicial. La dosis diaria de 3 mg de liraglutida proporcionó exposiciones sistémicas adecuadas sobre el rango de pesos de 60 a 234 kg cuya respuesta a la exposición se evaluó en los ensayos clínicos. No se estudió la exposición a liraglutida en pacientes con un peso corporal superior a los 234 kg. Insuficiencia hepática: Se evaluó la farmacocinética de liraglutida en pacientes con diversos grados de insuficiencia hepática en un ensayo de dosis única (0.75 mg). La exposición a liraglutida disminuyó un 13-23% en pacientes con insuficiencia hepática de leve a moderada en comparación con los sujetos sanos. La exposición fue significativamente menor (44%) en pacientes con insuficiencia hepática grave (puntuación Child Pugh >9). Insuficiencia renal: La exposición a liraglutida disminuyó en pacientes con insuficiencia renal en comparación con los individuos con una función renal normal en un ensayo de dosis única (0.75 mg). La exposición a liraglutida disminuyó un 33%, un 14%, un 27% y un 26%, respectivamente, en pacientes con insuficiencia renal leve (clearance de creatinina CrCl 50-80 ml/min), moderada (CrCl 30-50 ml/min) y grave (CrCl <30 ml/min) y con enfermedad renal en etapa terminal con necesidad de diálisis. Población pediátrica: No se ha estudiado el tratamiento con Saxenda® en pacientes pediátricos. Datos preclínicos sobre seguridad: Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas o genotoxicidad. Se observaron tumores no letales en células C de tiroides en estudios de carcinogenicidad de 2 años de duración en ratas y ratones. En ratas no se ha observado el nivel sin efecto adverso observado (sigla NOAEL, en inglés). Estos tumores no se observaron en monos tratados durante 20 meses. Estos hallazgos en roedores están provocados por un mecanismo específico no genotóxico mediado por el receptor GLP-1 al que los roedores son especialmente sensibles. La relevancia en humanos es probablemente baja pero no se puede excluir completamente. No se ha detectado ningún otro tumor relacionado con el tratamiento. Los estudios en animales no sugieren efectos perjudiciales directos en términos de fertilidad, pero sí un leve aumento de las muertes embrionarias tempranas a la dosis más alta. La administración de liraglutida durante el período intermedio de gestación provocó una reducción en el peso de la madre y en el crecimiento del feto con efectos no claros sobre las costillas en ratas y en la variación esquelética en el conejo. El crecimiento neonatal se redujo en el caso de las ratas durante su exposición a liraglutida y continuó durante el período de destete en el grupo de dosis elevada. Se desconoce si la disminución en el crecimiento de las crías se debe a una reducción en la ingesta de leche debido a un efecto directo del GLP-1 o a una reducción de la producción de leche materna a causa de una disminución de la ingesta calórica.
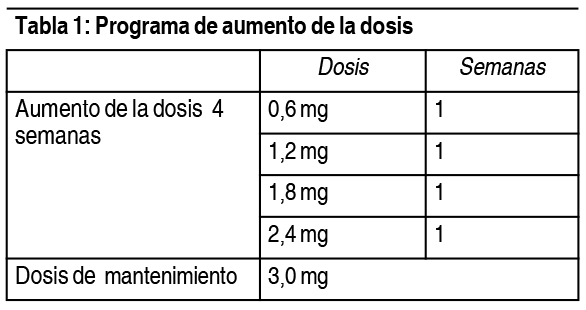
- Posología: La dosis inicial es de 0.6 mg al día. La dosis se debe aumentar hasta 3.0 mg al día en incrementos de 0.6 mg en intervalos de al menos 1 semana para que mejore la tolerancia gastrointestinal (ver tabla 1). Si el paciente no tolera un aumento de la dosis durante 2 semanas consecutivas, se debe considerar interrumpir el tratamiento. No se recomiendan dosis diarias superiores a 3.0 mg. Tabla 1. Programa de aumento de la dosis:
 El efecto del tratamiento se ha documentado solo para 1 año. Cada año se debe volver a evaluar si es necesario continuar con el tratamiento. Pacientes con diabetes mellitus tipo 2: Saxenda® no se debe utilizar en combinación con otro agonista del receptor de GLP-1. Cuando se inicia el tratamiento con Saxenda®, se debe considerar reducir la dosis de insulina o de secretagogos de la insulina (como sulfonilureas) que se administran de forma concomitante para reducir el riesgo de hipoglicemia. Poblaciones especiales: Pacientes de edad avanzada (≥65 años): No es necesario un ajuste de dosis en función de la edad. La experiencia terapéutica en pacientes de 75 años en adelante es limitada, por lo que no se recomienda su uso en estos pacientes (ver Advertencias y Precauciones especiales de empleo y propiedades farmacocinéticas). Pacientes con insuficiencia renal: No es necesario un ajuste de dosis en pacientes con insuficiencia renal leve o moderada (clearance de creatinina ≥30 ml/min). No se recomienda utilizar Saxenda® en pacientes con insuficiencia renal grave (clearance de creatinina <30 ml/min), incluidos los pacientes con enfermedad renal en etapa terminal (ver secciones Advertencias y Precauciones especiales de empleo, Efectos colaterales y Propiedades farmacocinéticas). Pacientes con insuficiencia hepática: No se recomienda ajustar la dosis en pacientes con insuficiencia hepática leve o moderada. No se recomienda utilizar Saxenda® en pacientes con insuficiencia hepática grave y se debe utilizar con precaución en pacientes con insuficiencia hepática leve o moderada (ver Advertencias y precauciones especiales de empleo y Propiedades farmacocinéticas). Población pediátrica: No se han establecido la seguridad y la eficacia de Saxenda® en niños y adolescentes menores de 18 años (ver Propiedades farmacodinámicas). No se dispone de datos. No se recomienda utilizar este medicamento en pacientes pediátricos. Método de administración: Saxenda® solo se debe administrar por vía subcutánea. No se debe administrar por vía intravenosa o intramuscular. Saxenda® se administra 1 vez al día en cualquier momento del día, con independencia de las comidas. Se debe inyectar en el abdomen, en el muslo o en la parte superior del brazo. Tanto el lugar de inyección como el momento de la administración se pueden modificar sin necesidad de ajustar la dosis. Sin embargo, es preferible que Saxenda® se inyecte alrededor de la misma hora del día, una vez que se haya elegido el momento más conveniente del día para ello. Si el paciente olvida inyectarse 1 dosis a su hora habitual y han transcurrido menos de 12 horas desde entonces, se debe inyectar la dosis lo antes posible. Si quedan menos de 12 horas para la siguiente dosis, el paciente no se debe inyectar la dosis olvidada y retomar el régimen de la dosis diaria habitual, es decir con la siguiente dosis programada. No se debe inyectar 1 dosis adicional ni aumentar la dosis para compensar la dosis olvidada. Para consultar más instrucciones sobre la administración, ver Precauciones especiales de eliminación y otras manipulaciones.
El efecto del tratamiento se ha documentado solo para 1 año. Cada año se debe volver a evaluar si es necesario continuar con el tratamiento. Pacientes con diabetes mellitus tipo 2: Saxenda® no se debe utilizar en combinación con otro agonista del receptor de GLP-1. Cuando se inicia el tratamiento con Saxenda®, se debe considerar reducir la dosis de insulina o de secretagogos de la insulina (como sulfonilureas) que se administran de forma concomitante para reducir el riesgo de hipoglicemia. Poblaciones especiales: Pacientes de edad avanzada (≥65 años): No es necesario un ajuste de dosis en función de la edad. La experiencia terapéutica en pacientes de 75 años en adelante es limitada, por lo que no se recomienda su uso en estos pacientes (ver Advertencias y Precauciones especiales de empleo y propiedades farmacocinéticas). Pacientes con insuficiencia renal: No es necesario un ajuste de dosis en pacientes con insuficiencia renal leve o moderada (clearance de creatinina ≥30 ml/min). No se recomienda utilizar Saxenda® en pacientes con insuficiencia renal grave (clearance de creatinina <30 ml/min), incluidos los pacientes con enfermedad renal en etapa terminal (ver secciones Advertencias y Precauciones especiales de empleo, Efectos colaterales y Propiedades farmacocinéticas). Pacientes con insuficiencia hepática: No se recomienda ajustar la dosis en pacientes con insuficiencia hepática leve o moderada. No se recomienda utilizar Saxenda® en pacientes con insuficiencia hepática grave y se debe utilizar con precaución en pacientes con insuficiencia hepática leve o moderada (ver Advertencias y precauciones especiales de empleo y Propiedades farmacocinéticas). Población pediátrica: No se han establecido la seguridad y la eficacia de Saxenda® en niños y adolescentes menores de 18 años (ver Propiedades farmacodinámicas). No se dispone de datos. No se recomienda utilizar este medicamento en pacientes pediátricos. Método de administración: Saxenda® solo se debe administrar por vía subcutánea. No se debe administrar por vía intravenosa o intramuscular. Saxenda® se administra 1 vez al día en cualquier momento del día, con independencia de las comidas. Se debe inyectar en el abdomen, en el muslo o en la parte superior del brazo. Tanto el lugar de inyección como el momento de la administración se pueden modificar sin necesidad de ajustar la dosis. Sin embargo, es preferible que Saxenda® se inyecte alrededor de la misma hora del día, una vez que se haya elegido el momento más conveniente del día para ello. Si el paciente olvida inyectarse 1 dosis a su hora habitual y han transcurrido menos de 12 horas desde entonces, se debe inyectar la dosis lo antes posible. Si quedan menos de 12 horas para la siguiente dosis, el paciente no se debe inyectar la dosis olvidada y retomar el régimen de la dosis diaria habitual, es decir con la siguiente dosis programada. No se debe inyectar 1 dosis adicional ni aumentar la dosis para compensar la dosis olvidada. Para consultar más instrucciones sobre la administración, ver Precauciones especiales de eliminación y otras manipulaciones. - Modo de Empleo: Precauciones especiales de eliminación y otras manipulaciones: La solución no se debe utilizar si no tiene un aspecto transparente e incoloro o casi incoloro. Saxenda® no se debe utilizar si se ha congelado. El dispositivo está diseñado para su utilización con agujas desechables NovoFine® o NovoTwist® de hasta 8 mm de longitud y tan delgadas como 32G. Las agujas no están incluidas en el envase. Se debe indicar al paciente que deseche la aguja después de cada inyección y que conserve el dispositivo sin la aguja puesta. De esta forma se evita contaminación, infecciones y la pérdida de producto. Así también se garantiza que la dosificación es precisa. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se deberá realizar de acuerdo con la normativa local. Instrucciones sobre cómo utilizar Saxenda® 6 mg/ml solución inyectable en dispositivo prellenado: Por favor lea atentamente estas instrucciones antes de utilizar su dispositivo prellenado Saxenda®. No utilice el dispositivo sin haber recibido la formación adecuada de su médico o enfermera. Empiece comprobando el dispositivo para asegurarse de que contiene Saxenda® 6 mg/ml y después observe las ilustraciones para familiarizarse con las distintas partes del dispositivo y la aguja. Si es ciego o tiene visión reducida y no puede leer el contador de dosis del dispositivo, no utilice este dispositivo sin ayuda. Busque la ayuda de una persona que vea bien y esté entrenada en el uso del dispositivo prellenado Saxenda®. Su dispositivo es un dispositivo prellenado dosificador. Contiene 18 mg de liraglutida, y administra dosis de 0.6 mg; 1.2 mg; 1.8 mg; 2.4 mg y 3.0 mg. El dispositivo está diseñado para utilizarse con agujas desechables NovoFine® o NovoTwist® de hasta 8 mm de longitud y tan delgadas como 32G. Las agujas no están incluidas en el envase. Información importante: Preste especial atención a estas notas porque son importantes para el uso seguro del dispositivo.
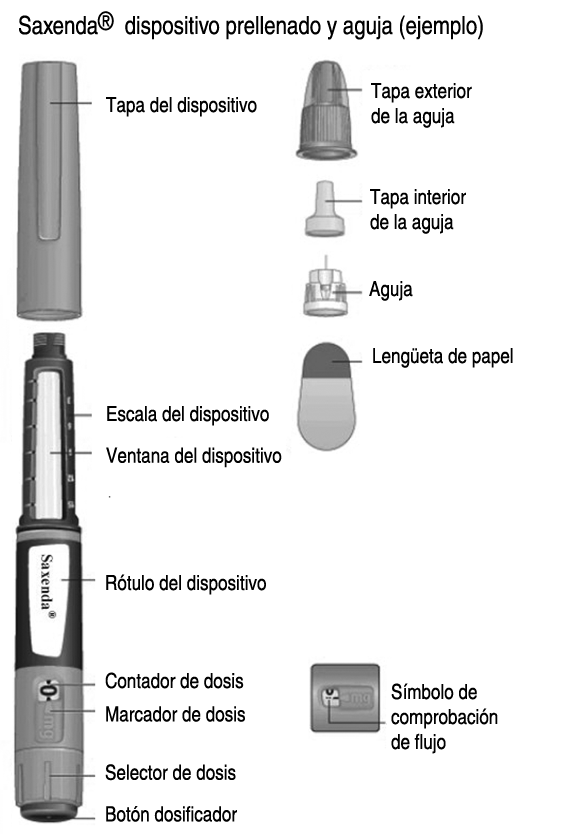 1.Preparación del dispositivo con una aguja nueva: A. 1. Compruebe el nombre y el color de la etiqueta de su dispositivo para asegurarse de que contiene Saxenda®. Esto es especialmente importante si utiliza más de 1 tipo de medicamento inyectable. El uso de un medicamento equivocado puede ser perjudicial para su salud. B. 1. Retire la tapa del dispositivo. Compruebe que la solución del dispositivo tiene un aspecto transparente e incoloro. Mire a través de la ventana del dispositivo. Si la solución tiene un aspecto turbio, no utilice el dispositivo. C. 1. Tome una aguja nueva y retire la lengüeta de papel. D. 1. Coloque la aguja recta en el dispositivo. Enrósquela hasta que quede apretada. E. 1. Retire la tapa exterior de la aguja y consérvela para más tarde. La necesitará después de la inyección para retirar la aguja del dispositivo de forma segura. F. 1. Retire la tapa interior de la aguja y deséchela. Si intenta volver a colocarla, puede pincharse accidentalmente con la aguja. Puede aparecer una gota de solución en la punta de la aguja. Esto es normal, pero a pesar de ello debe comprobar el flujo si utiliza un dispositivo nuevo por primera vez. No coloque una aguja nueva en el dispositivo hasta que esté listo para colocarse la inyección. Utilice siempre una aguja nueva para cada inyección. De esta forma puede evitar el atasco de las agujas, contaminación, infección e inexactitud de dosificación. Nunca utilice una aguja doblada o dañada.
1.Preparación del dispositivo con una aguja nueva: A. 1. Compruebe el nombre y el color de la etiqueta de su dispositivo para asegurarse de que contiene Saxenda®. Esto es especialmente importante si utiliza más de 1 tipo de medicamento inyectable. El uso de un medicamento equivocado puede ser perjudicial para su salud. B. 1. Retire la tapa del dispositivo. Compruebe que la solución del dispositivo tiene un aspecto transparente e incoloro. Mire a través de la ventana del dispositivo. Si la solución tiene un aspecto turbio, no utilice el dispositivo. C. 1. Tome una aguja nueva y retire la lengüeta de papel. D. 1. Coloque la aguja recta en el dispositivo. Enrósquela hasta que quede apretada. E. 1. Retire la tapa exterior de la aguja y consérvela para más tarde. La necesitará después de la inyección para retirar la aguja del dispositivo de forma segura. F. 1. Retire la tapa interior de la aguja y deséchela. Si intenta volver a colocarla, puede pincharse accidentalmente con la aguja. Puede aparecer una gota de solución en la punta de la aguja. Esto es normal, pero a pesar de ello debe comprobar el flujo si utiliza un dispositivo nuevo por primera vez. No coloque una aguja nueva en el dispositivo hasta que esté listo para colocarse la inyección. Utilice siempre una aguja nueva para cada inyección. De esta forma puede evitar el atasco de las agujas, contaminación, infección e inexactitud de dosificación. Nunca utilice una aguja doblada o dañada.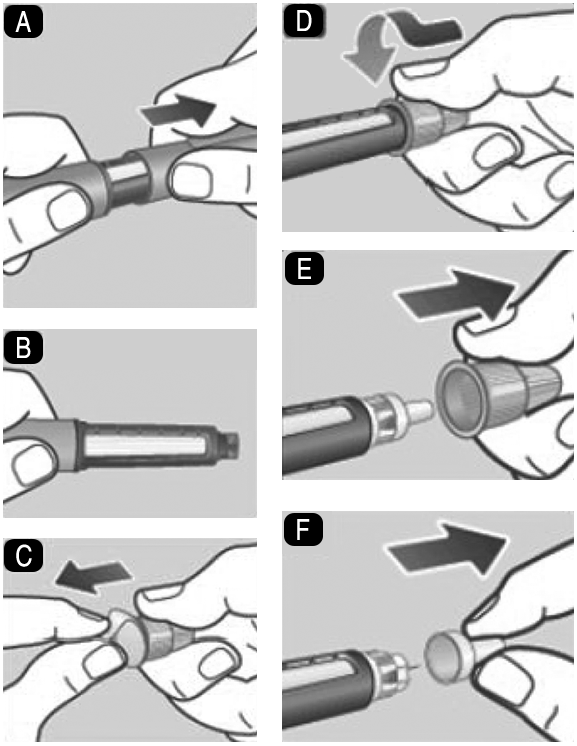 2. Comprobación del flujo: A. 2. Compruebe el flujo antes de la primera inyección con cada dispositivo nuevo. Si su dispositivo está ya en uso, vaya al paso 3 Selección de la dosis. Gire el selector de dosis hasta que aparezca el símbolo de comprobación de flujo en el cortador de dosis. B. 2. Sujete el dispositivo con la aguja apuntando hacia arriba. Presione y mantenga presionado el botón dosificador hasta que el contador de dosis vuelva a 0. El 0 debe quedar alineado con el marcador de dosis. Debe aparecer una gota de solución en la punta de la aguja. Puede que una pequeña gota se quede en la punta de la aguja, pero no se inyectará. Si no aparece ninguna gota, repita el paso 2 Comprobación del flujo, hasta 6 veces. Si sigue sin aparecer una gota, cambie la aguja y repita el paso 2 Comprobación del flujo, una vez más. Si, a pesar de todo, no aparece una gota, deseche el dispositivo y utilice uno nuevo. Asegúrese siempre de que aparezca una gota en la punta de la aguja antes de utilizar un dispositivo nuevo por primera vez. Así se asegura de que la solución fluye. Si no aparece una gota, no se inyectará medicamento, aunque el contador de dosis se mueva. Esto puede indicar que la aguja está bloqueada o dañada. Si no comprueba el flujo antes de la primera inyección con cada dispositivo nuevo, es posible que no se administre la dosis prescrita y que Saxenda® no produzca el efecto deseado.
2. Comprobación del flujo: A. 2. Compruebe el flujo antes de la primera inyección con cada dispositivo nuevo. Si su dispositivo está ya en uso, vaya al paso 3 Selección de la dosis. Gire el selector de dosis hasta que aparezca el símbolo de comprobación de flujo en el cortador de dosis. B. 2. Sujete el dispositivo con la aguja apuntando hacia arriba. Presione y mantenga presionado el botón dosificador hasta que el contador de dosis vuelva a 0. El 0 debe quedar alineado con el marcador de dosis. Debe aparecer una gota de solución en la punta de la aguja. Puede que una pequeña gota se quede en la punta de la aguja, pero no se inyectará. Si no aparece ninguna gota, repita el paso 2 Comprobación del flujo, hasta 6 veces. Si sigue sin aparecer una gota, cambie la aguja y repita el paso 2 Comprobación del flujo, una vez más. Si, a pesar de todo, no aparece una gota, deseche el dispositivo y utilice uno nuevo. Asegúrese siempre de que aparezca una gota en la punta de la aguja antes de utilizar un dispositivo nuevo por primera vez. Así se asegura de que la solución fluye. Si no aparece una gota, no se inyectará medicamento, aunque el contador de dosis se mueva. Esto puede indicar que la aguja está bloqueada o dañada. Si no comprueba el flujo antes de la primera inyección con cada dispositivo nuevo, es posible que no se administre la dosis prescrita y que Saxenda® no produzca el efecto deseado.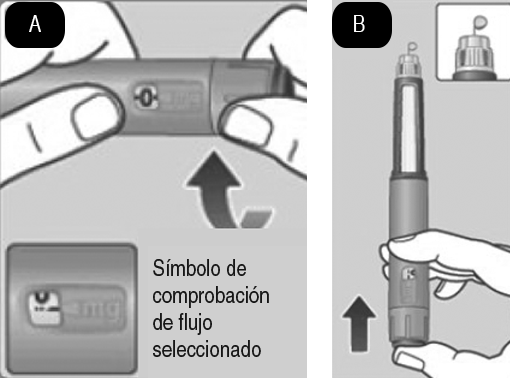
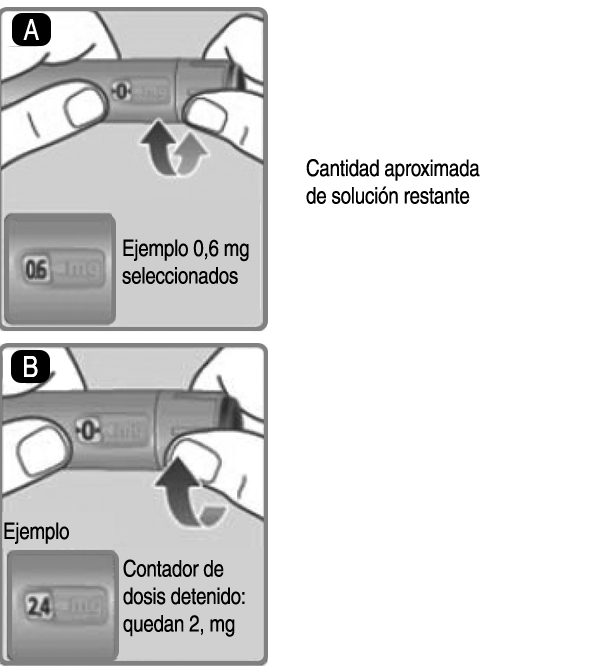
- 3. Selección de la dosis: A. 3. Gire el selector de dosis hasta que aparezca la dosis en el contador de dosis (0.6 mg; 1.2 mg; 1.8 mg; 2.4 mg o 3.00 mg). Si ha seleccionado una dosis incorrecta, puede girar el selector de dosis hacia delante o hacia atrás para seleccionar la dosis correcta. El dispositivo puede seleccionar hasta un máximo de 3.0 mg. El selector de dosis cambia la dosis. Solamente el contador de dosis y el marcador de dosis muestran cuántos mg ha seleccionado en cada administración. Puede seleccionar hasta 3.0 mg por dosis. Cuando el dispositivo contiene menos de 3.0 mg, el contador de dosis se detiene antes de que aparezca 3.0 mg. El selector de dosis hace clic de forma diferente cuando se gira hacia delante, hacia atrás o se pasa del número de mg que quedan. No cuente los clics del dispositivo. Antes de inyectarse el medicamento, utilice siempre el contador de dosis y el marcador de dosis para ver cuántos mg ha seleccionado. No cuente los clics del dispositivo. No utilice la escala del dispositivo, ya que solo muestra la cantidad aproximada de solución que queda en el dispositivo. Con el selector de dosis sólo deben seleccionarse dosis de 0.6 mg; 1.2 mg; 1.8 mg; 2.4 mg o 3.0 mg. La dosis seleccionada tiene que estar alineada de manera exacta con el marcador de dosis inyectada es la correcta. B. 3. Cuánta solución queda: La escala del dispositivo muestra la cantidad aproximada de solución que queda en el dispositivo. C. 3. Para ver cuánta solución queda exactamente, utilice el contador de dosis: Gire el selector de dosis hasta que el contador de dosis se detenga. Si muestra 3.0, significa que quedan al menos 3.0 mg en el dispositivo. Si el contador de dosis se detiene antes de 3.0 mg, significa que no queda suficiente solución para una dosis completa de 3.0 mg. Si necesita más medicamento del que queda en el dispositivo: Si el médico o enfermera lo aconsejan y le han enseñado a hacerlo, puede dividir la dosis entre el dispositivo en uso y uno nuevo. Utilice una calculadora para planificar la dosis según le haya indicado su médico o enfermera. Tenga mucho cuidado de hacer el cálculo correctamente. Si no está seguro de cómo dividir la dosis utilizando 2 dispositivos, seleccione e inyéctese la dosis que necesita con un dispositivo nuevo.
 4. Inyección de la dosis: A. 4. Inserte la aguja bajo la piel tal como le ha enseñado su médico o enfermera. Compruebe que puede ver el contador de dosis. No lo tape con los dedos. Esto podría interrumpir la inyección. B. 4.Presione y mantenga presionado el botón dosificador hasta que el contador de dosis indique 0. El 0 debe quedar alineado con el marcador de dosis. En ese momento puede que oiga o sienta un clic. C. 4. Mantenga la aguja bajo la piel después de que el contador de dosis haya vuelto a 0 y cuente lentamente hasta 6. Si retira antes la aguja, puede que vea salir solución desde la punta de la aguja. Si esto es así, significa que no se habrá administrado la dosis completa. D. 4. Retire la aguja de la piel. Si aparece sangre en el lugar de inyección, presione ligeramente. No frote la zona. Puede aparecer una gota de solución en la punta de la aguja después de la inyección. Esto es normal y no afecta a la dosis. Observe siempre el contador de dosis para saber cuántos mg inyecta. Presione y mantenga presionado el botón dosificador hasta que el contador de dosis indique 0. Cómo detectar si la aguja está bloqueada o dañada: Si no aparece el 0 en el contador de dosis después de presionar continuamente el botón dosificador, puede que haya usado una aguja bloqueada o dañada. En este caso, no habrá recibido nada de medicamento, incluso aunque el contador de dosis se haya movido desde la dosis original que seleccionó. Qué hacer si la aguja está bloqueada: Cambie la aguja tal como se describe en el paso 5. Después de la inyección, y repita todos los pasos desde el paso 1 Preparación del dispositivo con una aguja nueva. Asegúrese de seleccionar la dosis completa que usted necesita. No toque nunca el contador de dosis mientras se está inyectando. Esto puede interrumpir la inyección.
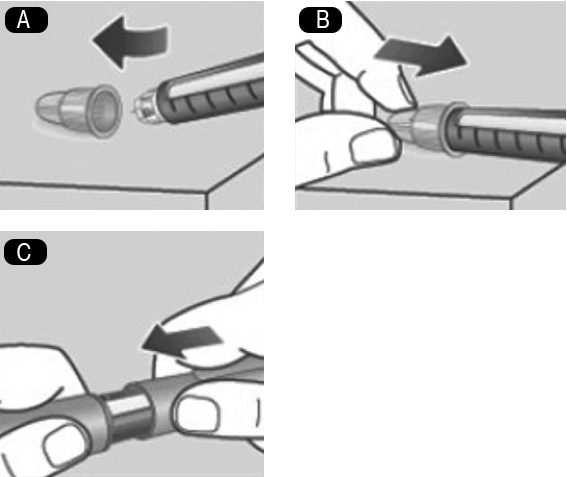
4. Inyección de la dosis: A. 4. Inserte la aguja bajo la piel tal como le ha enseñado su médico o enfermera. Compruebe que puede ver el contador de dosis. No lo tape con los dedos. Esto podría interrumpir la inyección. B. 4.Presione y mantenga presionado el botón dosificador hasta que el contador de dosis indique 0. El 0 debe quedar alineado con el marcador de dosis. En ese momento puede que oiga o sienta un clic. C. 4. Mantenga la aguja bajo la piel después de que el contador de dosis haya vuelto a 0 y cuente lentamente hasta 6. Si retira antes la aguja, puede que vea salir solución desde la punta de la aguja. Si esto es así, significa que no se habrá administrado la dosis completa. D. 4. Retire la aguja de la piel. Si aparece sangre en el lugar de inyección, presione ligeramente. No frote la zona. Puede aparecer una gota de solución en la punta de la aguja después de la inyección. Esto es normal y no afecta a la dosis. Observe siempre el contador de dosis para saber cuántos mg inyecta. Presione y mantenga presionado el botón dosificador hasta que el contador de dosis indique 0. Cómo detectar si la aguja está bloqueada o dañada: Si no aparece el 0 en el contador de dosis después de presionar continuamente el botón dosificador, puede que haya usado una aguja bloqueada o dañada. En este caso, no habrá recibido nada de medicamento, incluso aunque el contador de dosis se haya movido desde la dosis original que seleccionó. Qué hacer si la aguja está bloqueada: Cambie la aguja tal como se describe en el paso 5. Después de la inyección, y repita todos los pasos desde el paso 1 Preparación del dispositivo con una aguja nueva. Asegúrese de seleccionar la dosis completa que usted necesita. No toque nunca el contador de dosis mientras se está inyectando. Esto puede interrumpir la inyección.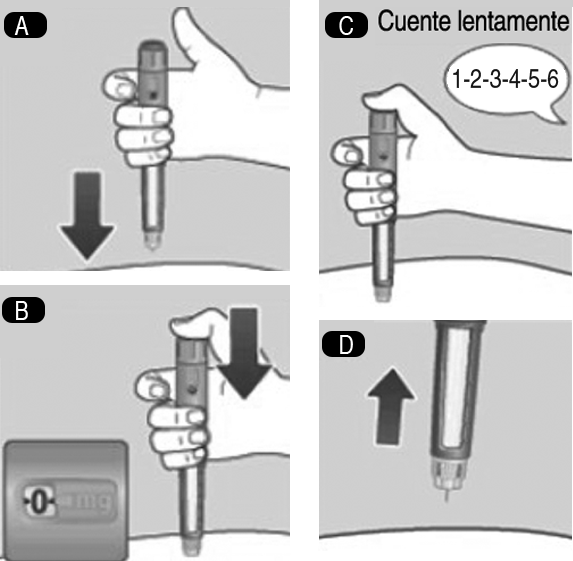 5. Después de la inyección: A. 5. Introduzca la punta de la aguja en su tapa exterior sobre una superficie plana, sin tocar la aguja ni la tapa exterior de la aguja. B. 5. Cuando la aguja esté cubierta, presione completamente y con cuidado la tapa exterior. Desenrosque la aguja y deséchela con cuidado. C. 5. Ponga la tapa en el dispositivo después de cada uso para proteger la solución de la luz. Deseche siempre la aguja después de cada inyección para asegurar que las inyecciones se administren correctamente y evitar que se atasquen las agujas. Si la aguja está atascada, no se inyectará medicamento. Cuando el dispositivo esté vacío, deséchelo sin la aguja puesta, siguiendo las instrucciones de su médico, enfermera, farmacéutico o de las autoridades locales. Nunca intente volver a colocar la tapa interior de la aguja. Podría pincharse con ella. Retire siempre la aguja del dispositivo después de cada inyección. De esta forma puede evitar que las agujas se atasquen, contaminación, infección, pérdida de solución y dosificación inexacta.
5. Después de la inyección: A. 5. Introduzca la punta de la aguja en su tapa exterior sobre una superficie plana, sin tocar la aguja ni la tapa exterior de la aguja. B. 5. Cuando la aguja esté cubierta, presione completamente y con cuidado la tapa exterior. Desenrosque la aguja y deséchela con cuidado. C. 5. Ponga la tapa en el dispositivo después de cada uso para proteger la solución de la luz. Deseche siempre la aguja después de cada inyección para asegurar que las inyecciones se administren correctamente y evitar que se atasquen las agujas. Si la aguja está atascada, no se inyectará medicamento. Cuando el dispositivo esté vacío, deséchelo sin la aguja puesta, siguiendo las instrucciones de su médico, enfermera, farmacéutico o de las autoridades locales. Nunca intente volver a colocar la tapa interior de la aguja. Podría pincharse con ella. Retire siempre la aguja del dispositivo después de cada inyección. De esta forma puede evitar que las agujas se atasquen, contaminación, infección, pérdida de solución y dosificación inexacta.
- Más información importante: Mantenga siempre el dispositivo y las agujas fuera de la vista y del alcance de otras personas, especialmente de los niños. Nunca comparta el dispositivo o las agujas con otras personas. Las personas que atienden a los pacientes deben tener mucho cuidado cuando manejen agujas usadas para evitar pinchazos accidentales e infecciones. Cuidados del dispositivo: No deje el dispositivo en el automóvil ni en otro lugar donde pueda calentarse o enfriarse en exceso. No se inyecte Saxenda® si se ha congelado. Si lo hace, no conseguirá el efecto deseado de este medicamento. No exponga el dispositivo al polvo, suciedad o líquidos. No lave, remoje ni lubrique el dispositivo. Si es necesario, límpiela con un paño humedecido con un detergente suave. Cuide que el dispositivo no se caiga ni golpee contra superficies duras. Si se le cae el dispositivo o sospecha que pueda tener un problema, coloque una aguja nueva y compruebe el flujo antes de inyectarse. No intente rellenar el dispositivo. Una vez vacío, se debe desechar. No intente reparar el dispositivo ni desmontarlo.
- Efectos Colaterales: Resumen del perfil de seguridad: El programa de desarrollo clínico de Saxenda® consta de 6 ensayos clínicos completos en los que han participado 5.813 pacientes obesos o pacientes con sobrepeso con al menos 1 comorbilidad relacionada con el peso. En general, las reacciones gastrointestinales fueron las reacciones adversas notificadas más frecuentemente durante el tratamiento con Saxenda® (ver Descripción de las reacciones adversas seleccionadas). Lista tabulada de reacciones adversas: En la tabla 2 se presentan las Reacciones Adversas notificadas en ensayos de fase 3 controlados a largo plazo. Las reacciones adversas figuran en la lista según el sistema de clasificación de órganos y la frecuencia. Las categorías de frecuencia se definen del siguiente modo: muy frecuentes (≥1/10); frecuentes (≥1/100 a <1/10); poco frecuentes (≥1/1.000 a <1/100); raras (≥1/10.000 a <1/1.000); muy raras (<1/10.000). Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Tabla 2. Reacciones adversas notificadas en estudios controlados de fase 3. Sistema de clasificación de órganos MedDRA: Trastornos del sistema inmunológico: Raras: Reacción anafiláctica. Trastornos del metabolismo y de la nutrición: Frecuentes: Hipoglicemia*. Poco frecuentes: Deshidratación. Trastornos psiquiátricos: Frecuentes: Insomnio**. Trastornos del sistema nervioso: Frecuentes: Mareo. Disgeusia. Trastornos cardíacos: Poco frecuentes: Taquicardia. Trastornos gastrointestinales: Muy frecuentes: Náuseas, vómitos, diarrea, estreñimiento. Frecuentes: Sequedad de boca, dispepsia, gastritis, enfermedad de reflujo gastroesofágico, dolor abdominal superior, flatulencia, eructos, distensión abdominal. Poco frecuentes: Pancreatitis***. Trastornos hepatobiliares: Frecuentes: Colelitiasis***. Poco frecuentes: Colecistitis***. Trastornos de la piel y del tejido subcutáneo: Poco frecuentes: Urticaria. Trastornos renales y urinarios: Raras: Falla renal aguda, insuficiencia renal. Trastornos generales y alteraciones en el lugar de administración: Frecuentes: Reacciones en el lugar de inyección, astenia, fatiga. Poco frecuentes: Malestar. *Se notificaron casos de hipoglicemia (según los síntomas indicados por los pacientes y no confirmados mediante mediciones de glucosa en sangre) en pacientes sin diabetes mellitus tipo 2 en tratamiento con Saxenda® en combinación con dieta y ejercicio. Para obtener más información, ver Descripción de las reacciones adversas seleccionadas. **El insomnio se produjo principalmente durante los 3 primeros meses de tratamiento. ***Ver Advertencias y precauciones especiales de empleo. Descripción de las reacciones adversas seleccionadas: Hipoglicemia en pacientes sin diabetes mellitus tipo 2: En ensayos clínicos realizados en pacientes obesos o con sobrepeso sin diabetes mellitus tipo 2 en tratamiento con Saxenda® en combinación con dieta y ejercicio, no se notificaron episodios hipoglicémicos graves (que requiriesen la asistencia de terceras personas). El 1.6% de los pacientes tratados con Saxenda® y el 1.1% de los pacientes tratados con placebo notificaron que presentaron síntomas de episodios hipoglicémicos; sin embargo, estos episodios no se confirmaron mediante mediciones de glucosa en sangre. La mayoría de los episodios fueron leves. Hipoglicemia en pacientes con diabetes mellitus tipo 2: En un ensayo clínico realizado en pacientes obesos o con sobrepeso con diabetes mellitus tipo 2 en tratamiento con Saxenda® en combinación con dieta y ejercicio, se notificó hipoglicemia grave (que requirió la asistencia de terceras personas) en el 0.7% de los pacientes tratados con Saxenda® y solamente en pacientes tratados de forma concomitante con sulfonilurea. Además, se documentaron casos de hipoglicemia sintomática en el 43.6% de los pacientes tratados con Saxenda® y en el 27.3% de los pacientes tratados con placebo. Entre los pacientes que no se trataron con sulfonilurea de forma concomitante, se documentaron episodios de hipoglicemia sintomática (definidos como glucosa en plasma ≤3.9 mmol/l acompañados de síntomas) en el 15.7% de los pacientes tratados con Saxenda® y el 7.6% de los pacientes tratados con placebo. Reacciones adversas gastrointestinales: La mayoría de los episodios gastrointestinales fueron de leves a moderados, transitorios y no conllevaron la interrupción del tratamiento. Las reacciones generalmente sucedieron durante las primeras semanas y disminuyeron una vez transcurridos algunos días o semanas de tratamiento continuado. Los pacientes de 65 años de edad en adelante pueden experimentar más efectos gastrointestinales al ser tratados con Saxenda®. Los pacientes con insuficiencia renal leve o moderada (clearance de creatinina ≥30 ml/min) pueden experimentar más efectos gastrointestinales al ser tratados con Saxenda®. Falla renal aguda: En pacientes tratados con agonistas del receptor de GLP-1 se han notificado casos de falla renal aguda. La mayoría de los casos notificados se produjeron en pacientes que habían experimentado náuseas, vómitos o diarrea con la consiguiente disminución del volumen (ver Advertencias y precauciones especiales de empleo). Reacciones alérgicas: Durante la comercialización de liraglutida, se han notificado pocos casos de reacciones anafilácticas con síntomas tales como hipotensión, palpitaciones, disnea y edema. Las reacciones anafilácticas pueden ser potencialmente mortales. Ante la sospecha de una reacción anafiláctica, se debe interrumpir el tratamiento con liraglutida y este no se debe reanudar (ver Contraindicaciones). Reacciones en el lugar de inyección: En pacientes tratados con Saxenda® se han notificado reacciones en el lugar de inyección. Estas reacciones fueron por lo general leves y transitorias y la mayoría desaparecieron durante el tratamiento continuado. Taquicardia: En ensayos clínicos se notificó taquicardia en el 0.6% de los pacientes tratados con Saxenda® y en el 0.1% de los pacientes tratados con placebo. La mayoría de los episodios fueron leves o moderados. Se trató de episodios aislados que en su mayoría se resolvieron durante el tratamiento continuado con Saxenda®.
- Contraindicaciones: Hipersensibilidad a liraglutida o a alguno de los excipientes.
- Advertencias: Fertilidad, embarazo y lactancia: Embarazo: Los datos que existen sobre la utilización de liraglutida en mujeres embarazadas son limitados. Los estudios en animales han mostrado toxicidad para la reproducción (ver Datos preclínicos sobre seguridad). Se desconoce el riesgo en seres humanos. No se debe administrar liraglutida durante el embarazo. Se debe interrumpir el tratamiento con liraglutida en caso de que una paciente desee quedarse embarazada o si se produce un embarazo. Lactancia: Se desconoce si liraglutida se excreta en la leche materna. Estudios realizados en animales han mostrado que la transferencia a la leche de liraglutida y metabolitos de estrecha relación estructural es baja. Estudios no clínicos han mostrado una reducción en el crecimiento neonatal relacionada con el tratamiento en crías de rata en período de lactancia (ver Datos preclínicos sobre seguridad). Dada la falta de experiencia, no se debe usar Saxenda® durante el período de lactancia. Fertilidad: Los estudios en animales no han revelado efectos nocivos relacionados con la fertilidad, aparte de una ligera disminución en el número de implantes vivos (ver Datos preclínicos sobre seguridad). Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de Saxenda® sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
- Precauciones: Advertencias y precauciones especiales de empleo: En pacientes con diabetes mellitus no se debe utilizar liraglutida como sustituto de la insulina. La experiencia en pacientes con insuficiencia cardíaca congestiva de clase I y II según la New York Heart Association (NYHA) es limitada y, por lo tanto, liraglutida se debe utilizar con precaución. No existe experiencia en pacientes con insuficiencia cardíaca congestiva de clase III y IV según la NYHA y, por lo tanto, liraglutida no se recomienda en estos pacientes. No se han establecido la seguridad y la eficacia de liraglutida para controlar el peso en pacientes: De 75 años de edad en adelante. Tratados con otros productos para controlar el peso, con obesidad debida a trastornos endocrinos o alimenticios o al tratamiento con medicamentos que pueden provocar aumento de peso. Con insuficiencia renal grave. Con insuficiencia hepática grave. No se recomienda el uso en estos pacientes (ver Posología). Como liraglutida no se ha estudiado para controlar el peso en sujetos con insuficiencia hepática leve o moderada, se debe utilizar con precaución en estos pacientes (ver Posología y Propiedades farmacocinéticas) La experiencia en pacientes con enfermedad inflamatoria intestinal y gastroparesia diabética es limitada. No se recomienda el uso de liraglutida en estos pacientes ya que se asocia a reacciones adversas gastrointestinales transitorias, como náuseas, vómitos y diarrea. Pancreatitis: El uso de agonistas del receptor de GLP-1 se ha asociado con el riesgo de desarrollar pancreatitis aguda. Se han notificado pocos casos de pancreatitis aguda con liraglutida. Se debe informar a los pacientes de los síntomas característicos de la pancreatitis aguda. Ante la sospecha de pancreatitis, se debe interrumpir el tratamiento con liraglutida, y este no se debe reanudar si se confirma la pancreatitis aguda. Se debe extremar la precaución en pacientes con antecedentes de pancreatitis. Colelitiasis y colecistitis: En ensayos clínicos sobre control del peso, se ha observado una mayor tasa de colelitiasis y colecistitis en los pacientes tratados con liraglutida que en los pacientes tratados con placebo. El hecho de que la pérdida importante de peso puede aumentar el riesgo de colelitiasis y, por consiguiente de colecistitis, solo explicó parcialmente la mayor tasa con liraglutida. La colelitiasis y la colecistitis pueden requerir hospitalización y colecistectomía. Se debe informar a los pacientes de los síntomas característicos de la colelitiasis y la colecistitis. Enfermedad tiroidea: En ensayos clínicos llevados a cabo en pacientes con diabetes tipo 2, se han notificado acontecimientos adversos tiroideos, que incluyen aumento de calcitonina en sangre, bocio y neoplasia tiroidea, especialmente en pacientes con enfermedad tiroidea preexistente. En los ensayos clínicos de control del peso también se han observado casos de aumento de calcitonina en sangre. Por tanto, liraglutida se debe utilizar con precaución en pacientes con enfermedad tiroidea. Frecuencia cardíaca: En ensayos clínicos se ha observado que liraglutida produce un aumento de la frecuencia cardíaca (ver Propiedades farmacodinámicas). No está clara la importancia clínica del aumento de la frecuencia cardíaca con el tratamiento de liraglutida, especialmente en pacientes con cardiopatía o enfermedad cerebrovascular, como resultado de la exposición limitada de estos pacientes en los ensayos clínicos. La frecuencia cardíaca se debe controlar de forma periódica de acuerdo con la práctica clínica habitual. Se debe informar a los pacientes de los síntomas del aumento de la frecuencia cardíaca (palpitaciones o sensación de aceleración del pulso en reposo). El tratamiento con liraglutida se debe interrumpir en pacientes que experimenten un incremento sostenido clínicamente significativo de la frecuencia cardíaca en reposo. Deshidratación: Se han notificado signos y síntomas de deshidratación que incluyen insuficiencia renal y fallo renal agudo en pacientes en tratamiento con agonistas del receptor de GLP-1. Se debe advertir a los pacientes en tratamiento con liraglutida de que existe un riesgo potencial de deshidratación relacionado con los efectos adversos gastrointestinales y de que tomen precauciones para evitar la pérdida de líquidos. Hipoglicemia en pacientes con diabetes mellitus tipo 2: Los pacientes con diabetes mellitus tipo 2 a los que se les administra liraglutida en combinación con una sulfonilurea podrían presentar un riesgo mayor de hipoglicemia. Es posible disminuir el riesgo de hipoglicemia reduciendo la dosis de sulfonilurea. No se ha estudiado la adición de Saxenda® en pacientes tratados con insulina.
- Interacciones Medicamentosas: Interacción con otros medicamentos y otras formas de interacción: In vitro, liraglutida ha demostrado un potencial muy bajo de estar implicada en interacciones farmacocinéticas con otras sustancias activas relacionadas con el citocromo P450 (CYP) y la unión a proteínas plasmáticas. El leve retraso en el vaciamiento gástrico asociado a liraglutida puede influir en la absorción de medicamentos orales administrados de forma concomitante. Los estudios de interacción no han demostrado ningún retraso clínicamente significativo en la absorción y, por lo tanto, no se requiere ajuste de dosis. Se han realizado estudios de interacciones con 1.8 mg de liraglutida. El efecto sobre la tasa de vaciamiento gástrico fue equivalente para 1.8 mg y 3.0 mg de liraglutida, (ABC0-300 min de paracetamol). Pocos pacientes tratados con liraglutida notificaron al menos un episodio de diarrea grave. La diarrea puede influir en la absorción de medicamentos de administración oral concomitante. Warfarina y otros derivados de la cumarina: No se ha realizado estudio de interacción. No se puede excluir una interacción clínicamente significativa con principios activos con escasa solubilidad o índice terapéutico estrecho, tales como la warfarina. Al inicio del tratamiento con liraglutida en pacientes tratados con warfarina u otros derivados de la cumarina, se recomienda un control de la Razón Internacional Normalizada (sigla INR, en inglés) más frecuente. Paracetamol (acetaminofeno): Liraglutida no modificó la exposición total del paracetamol tras la administración de una dosis única de 1.000 mg. Se produjo una disminución del 31% en la Cmáx de paracetamol y un retraso en el tmáx medio de hasta 15 min. No es necesario un ajuste de dosis en el uso concomitante de paracetamol. Atorvastatina: Liraglutida no modificó la exposición total de atorvastatina tras la administración de una dosis única de 40 mg de atorvastatina. Por lo tanto, no es necesario un ajuste de dosis de atorvastatina cuando se administra con liraglutida. Se produjo una disminución del 38% en la Cmáx de atorvastatina y el tmáx medio se retrasó de 1 a 3 horas con liraglutida. Griseofulvina: Liraglutida no modificó la exposición total de griseofulvina tras la administración de una dosis única de 500 mg de griseofulvina. Se produjo un aumento del 37% en la Cmáx de griseofulvina y el tmáx medio permaneció inalterado. No es necesario un ajuste de dosis de griseofulvina y otros componentes de baja solubilidad y alta permeabilidad. Digoxina: La administración de una única dosis de 1 mg de digoxina con liraglutida produjo una reducción en la ABC de digoxina de un 16%; la Cmáx disminuyó un 31%. Se produjo un retraso en el tmáx medio de la digoxina de 1 a 1.5 horas. No es necesario un ajuste de dosis de digoxina en base a estos resultados. Lisinopril: La administración de una única dosis de 20 mg de lisinopril con liraglutida mostró una reducción en la ABC de lisinopril de un 15%; la Cmáx disminuyó un 27%. Se produjo un retraso en el tmáx medio del lisinopril que pasó de 6 a 8 horas con liraglutida. No es necesario un ajuste de dosis de lisinopril en base a estos resultados. Anticonceptivos orales: Tras la administración de una única dosis de un medicamento anticonceptivo oral, liraglutida disminuyó la Cmáx de etinilestradiol y levonorgestrel un 12% y un 13% respectivamente. Se produjo un retraso en el tmáx de alrededor de 1.5 horas con liraglutida para ambos compuestos. No se observó ningún efecto clínicamente significativo sobre la exposición total ni al etinilestradiol ni al levonorgestrel. Se prevé, por lo tanto, que el efecto anticonceptivo permanezca inalterado cuando se administran de forma conjunta con liraglutida.
- Sobredosificación: Durante los ensayos clínicos y el uso de liraglutida tras su comercialización, se han notificado casos de sobredosis de hasta 72 mg (24 veces la dosis recomendada para controlar el peso). Los casos notificados incluyeron náuseas y vómitos intensos, que son también los síntomas esperados de una sobredosis con liraglutida. Ninguna de las notificaciones incluyó hipoglicemia grave. Todos los pacientes se recuperaron sin complicaciones. En caso de sobredosis, se debe iniciar el tratamiento de soporte adecuado en función de los síntomas y signos clínicos del paciente. Se debe observar al paciente para detectar signos clínicos de deshidratación y se deben controlar sus niveles de glucosa en sangre.
- Incompatibilidades: Las sustancias añadidas a Saxenda® pueden provocar la degradación de liraglutida. En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
- Conservación: Conservar en refrigeración (2°C - 8°C). No congelar. Conservar lejos del elemento refrigerante. Después del primer uso: conservar por debajo de 30°C o en refrigeración (2°C - 8°C). El producto debe ser desechado 1 mes después de su primer uso. Conservar el dispositivo con la tapa puesta para protegerlo de la luz.
- Observaciones: Saxenda®, NovoFine® y NovoTwist® son marcas registradas de propiedad de Novo Nordisk A/S, Dinamarca. ©2015 Novo Nordisk A/S. Información importante en Chile: Registro ISP B-2626. Mayor información en www.ispch.cl. Importado por Novo Nordisk Farmacéutica Ltda., Av. Presidente Riesco 5335, Of. 504, Las Condes, Santiago y distribuido por Droguería Perilogistics Ltda., Rodrigo de Araya 1151, Macul, Santiago. Mantener fuera del alcance y de la vista de los niños. Venta bajo receta médica (R) en establecimientos tipo A. Uso bajo supervisión médica.
- Presentaciones: Naturaleza y contenido del envase: Cartucho (vidrio tipo 1) con un émbolo (bromobutilo) y un tapón (bromobutilo/poliisopreno) dentro de un dispositivo prellenado desechable multidosis hecho de polipropileno, poliacetal, policarbonato y acrilonitrilo butadieno estireno. Cada dispositivo contiene: 3 ml de solución pudiendo suministrar dosis de 0.6 mg; 1.2 mg; 1.8 mg; 2.4 mg y 3.0 mg. Envases con 1, 3 ó 5 dispositivos prellenados. Puede que solamente estén comercializados algunos tamaños de envases.


